Distrofias corneanas caracterizam-se por opacidades de caráter não inflamatório, em geral bilaterais e simétricas. Em grande parte são hereditárias, sendo em sua maioria de herança autossômica dominante. Em raros casos apresentam associação com doenças sistêmicas, e anatomicamente podem ser classificadas em: epiteliais, da camada de Bowman, estromais e endoteliais.
Continue a leitura e tire suas dúvidas!
Tipos de distrofia corneana: confira as principais
Distrofia de Meesman
Dentre as epiteliais, está a distrofia de Meesman (distrofia epitelial hereditária juvenil), que acomete pacientes desde a primeira década de vida. As mutações nos genes KRT3 e KRT12 causam degenerações citoplasmáticas nas células basais com a formação de cistos epiteliais que podem romper causando erosões corneanas, cursando com dor, fotofobia e lacrimejamento.
Distrofia de Cogan
A segunda distrofia epitelial é a distrofia de Cogan (Map-Dot-Fingerprint). É a distrofia corneana mais comum, sendo mais frequente em mulheres adultas, com acometimento do gene TGFBI e consequente distúrbio da ceratoepitelina. Apresenta-se com padrão característico de manchas acinzentadas, linhas e pseudocistos epiteliais, configuração típica que dá nome à distrofia. Em ambas, casos leves podem ser tratados com lubrificação e uso de lentes de contato terapêutica nas crises, sendo possível a realização de micropuntura ou ceratectomia fototerapêutica (PTK) nos casos avançados com opacidades graves, embora as recidivas sejam comuns.
Distrofia da cama de Bowman
As distrofias da camada de Bowman dividem-se em Reis-Bücklers e Thiel-Behnke. A primeira, também chamada de Bowman tipo I, costuma ter início precoce na infância, com erosões epiteliais recorrentes que tornam-se menos frequentes com a idade por conta do processo fibrótico instalado, levando à cicatrização irregular da córnea e consequentemente à baixa de visão. Tal processo fibroso com deposição de material granular pode ser visto na histopatologia com a coloração tricrômio de Masson. Já a Distrofia de Thiel-Behnke (Bowman tipo 2 ou Honeycomb) tem como característica a formação de opacidades centrais reticulares na camada de Bowman com aspecto em favo de mel, poupando inicialmente a periferia corneana. A diferenciação entre ambas pode ser difícil clinicamente, sendo o exame anatomopatológico ou microscopia confocal de grande valia. Seu tratamento é semelhante ao das distrofias epiteliais.
Mais profundamente na córnea, o estroma corneano pode ser acometido por diversas distrofias. A distrofia granular do tipo 1 (Groenow 1) acomete preferencialmente o estroma anterior com distribuição central em aspecto de flocos de neve, pelas opacidades por depósito hialino entremeadas com tecido corneano saudável. Dentre seus subtipos I e II, o primeiro apresenta-se mais precocemente na primeira infância com opacidades mais numerosas e maior potencial de causar baixa de visão. Já a Granular tipo II (de Avelino) apresenta características mistas entre a Distrofia Granular e a Distrofia Lattice, com depósitos hialinos e depósitos amiloides. A recidiva das lesões, mesmo após transplante, é frequente.
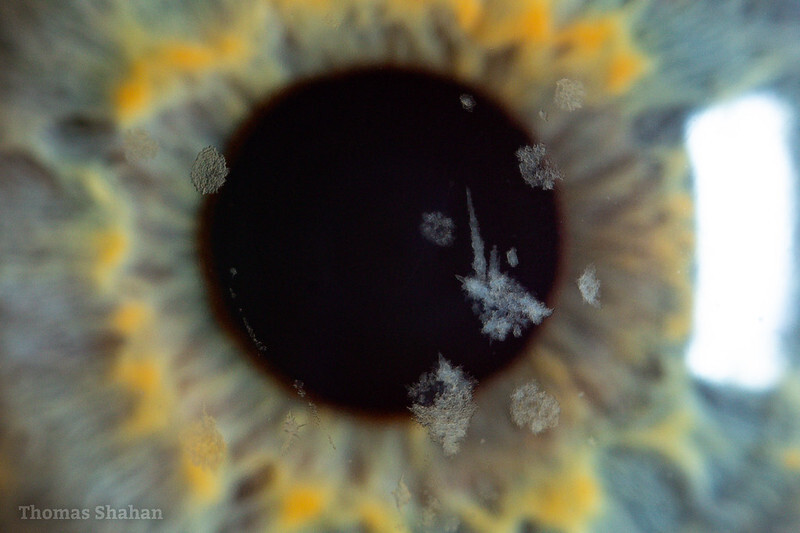
Distrofia granular (crédito: Thomas Shahan). https://www.flickr.com/photos/49580580@N02/51739845436.
Distrofia estromal
A distrofia estromal mais comum é a Lattice, que se divide em 3 subtipos, sendo o tipo III uma das raras entidades de herança autossômica recessiva. O tipo 1 (Biber-Haab-Dimmer) apresenta-se com linhas e opacidades puntiformes mais centrais, tipicamente poupando a periferia corneana. Inicia-se na primeira década de vida e apresenta erosões recorrentes com frequência. Já o tipo II (Meretoja) tem como características o início mais tardio entre a segunda e terceira décadas de vida e sua associação com amiloidose. Em geral as lesões apresentam-se a partir da periferia corneana, ao contrário do primeiro subtipo. Já no tipo III as linhas estromais são tipicamente mais grosseiras, enquanto as erosões são incomuns. Classicamente, a distrofia Lattice apresenta depósitos amiloides no estroma anterior que podem ser vistos pela birrefringência à luz polarizada, além de corarem-se com a coloração vermelho do congo.
Dentre as estromais, a distrofia Macular (Ou Groenow II) tem o maior potencial de causar baixa visual, embora seja a mais rara. De herança autossômica recessiva e acometimento central que se espalha para a periferia e tecidos corneanos profundos, suas opacidades por depósito de glicosaminoglicanos se diferenciam do tipo granular pela ausência de tecido corneano saudável entre as lesões. Classicamente pode ser vista pela coloração Alcian Blue.
Ainda dentre as distrofias que acometem o estroma, destacam-se a distrofia cristalina de Schnyder, com acometimento precoce característico com lesões centrais que evoluem com formação de anel periférico por depósitos lipídicos que coram com Sudan-Black; a distrofia cristalina de Bietti, de herança recessiva, tipicamente com depósitos cristalinos na extrema periferia corneana e na retina; a distrofia gelatinosa em gota, de aparecimento no primeiro ano de vida, com protuberâncias circulares gelatinosas quepodem evoluir com neovascularização tardia e recidivas frequentes, sendo em algumas referências caracterizada como distrofia epitelio-estromal; a distrofia nebulosa de François, causada pelo desarranjo anatômico das lamelas de colágeno que denota lesões geométricas poligonais no estroma profundo corneano entremeadas com tecido sadio; a distrofia salpicada de Fleck, cursando com depósitos de glicosaminoglicanos e lipídios em todo o estroma; e a distrofia estromal congênita, de padrão simétrico, difuso, bilateral e não progressiva, já presente ao nascimento, evoluindo com baixa visual importante.
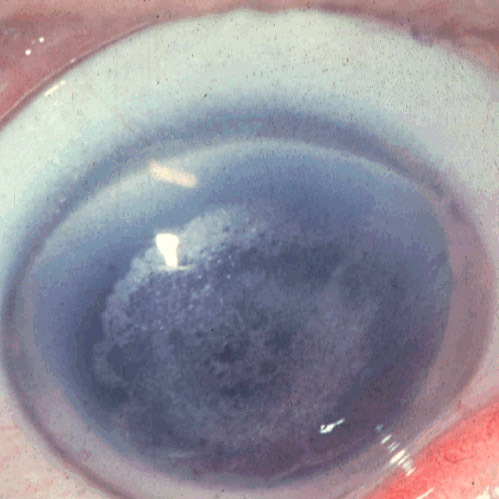
Distrofia cristalina de Schnyder
Distrofias com acometimento importante causando opacidades visuais consideráveis podem ser tratadas com transplante lamelar anterior superficial ou profundo, ou penetrante em casos graves.
Distrofias endoteliais
Por último, temos as distrofias endoteliais, que aumentam a chance de descompensação corneana pela quebra da barreira responsável pela deturgescência relativa. A córnea guttata não tem padrão de herança definido e caracteriza-se pela
produção anormal de colágeno pelo endotélio, denotando a presença de excrescências na Descemet com finos pigmentos em aspecto de metal batido na superfície posterior.
Distrofia de Fuchs
Já a distrofia de Fuchs tem herança autossômica dominante e é mais comum em mulheres acima de 40 anos, sendo bilateral, assimétrica e lentamente progressiva. Cursa com guttatas, inicialmente apresentando-se como baixa de visão pela manhã pelo edema estromal causado pelo aumento de ácido lático durante a noite com os olhos cerrados. Progride com edema estromal e superficial com bolhas epiteliais que podem romper e causar sintomas, tardiamente apresentando baixa visual importante pela fibrose e formação de pannus neovascular. Na microscopia especular evidenciam-se pleomorfismo, polimegatismo e diminuição da contagem endotelial. Assim como as outras afecções do endotélio, o tratamento pode ser feito com transplante lamelar posterior, ou em casos mais avançados opta-se pelo transplante penetrante ou micropuntura, se contraindicada a cirurgia.
Distrofia polimorfa
A distrofia polimorfa posterior tem relação com o gene PPCD e é bilateral, assimétrica e em geral apresenta baixa progressão das lesões, típicas do quadro: vesículas endoteliais, lesões estelares ou padrão geográfico de opacidade. É um importante diagnóstico diferencial das síndromes ICE (geralmente unilaterais) pela possibilidade de desenvolvimento de edema estromal, sinéquias anteriores e glaucoma. A fisiopatologia é descrita pela epitelização do endotélio corneano com queratinização.
Distrofia endoteliais
Última das distrofias endoteliais, a distrofia hereditária endotelial congênita (CHED) é uma condição rara bilateral, simétrica, não inflamatória, composta por opacidade corneana difusa acometendo a porção profunda de toda a córnea sem tecido saudável entreposto, com espessamento importante corneano. Divide-se em duas formas de acordo com sua herança genética: autossômica recessiva, não progressiva e presente desde o nascimento, com prognóstico visual ruim pela densidade das opacidades que impossibilitam o desenvolvimento visual e reflexo de fixação; e a forma autossômica dominante, com aparecimento mais tardio inicialmente com opacidades leves progressivas e melhor prognóstico visual.
O conhecimento acerca das principais distrofias corneanas possibilita o diagnóstico precoce e acompanhamento dos pacientes e de seus familiares, sendo possível a intervenção precoce se necessária, minimizando assim as chances de complicações como baixa visual definitiva, fibrose das camadas corneanas, necessidade de transplante penetrante e suas consequências, além de garantir melhor qualidade de vida aos pacientes.
Referência:
1. External Disease and Cornea – Basic and Clinical Science Course 2019-2020, American Academy of Ophthamology